|
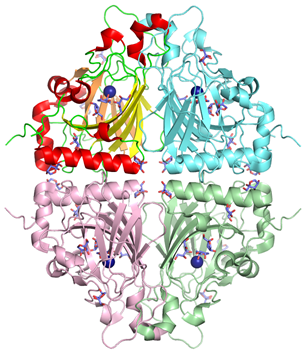
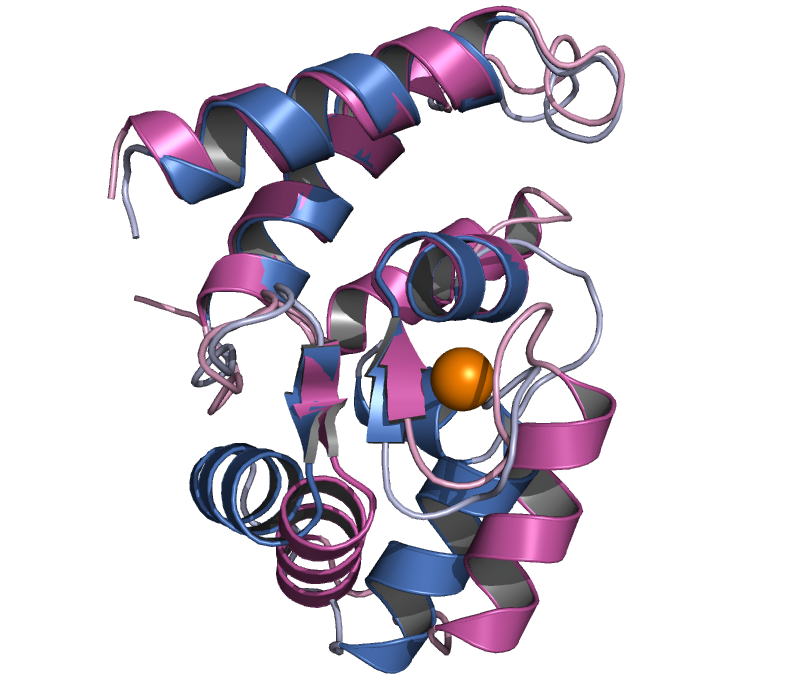
Acinetobacter
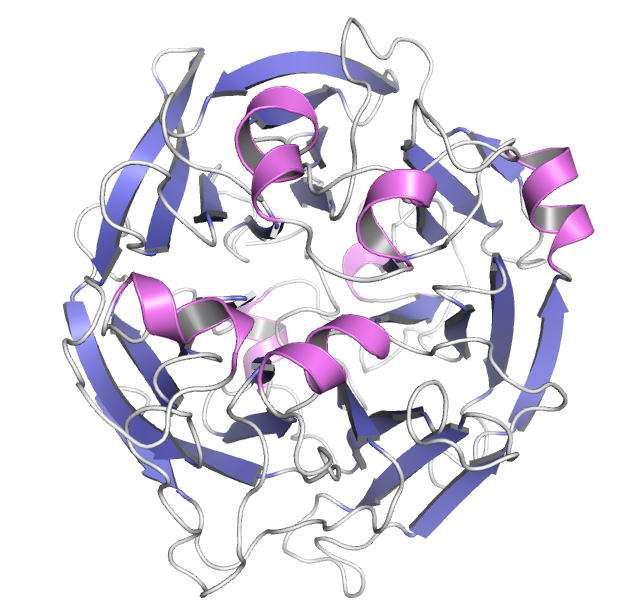
sp. DL-28 L-ribose isomerase (4Q0P) L-Ribose, a pentose, is not known to exist
in nature. Although organisms typically do not have a metabolic pathway that
uses L-ribose as a carbon source, prokaryotes use various sugars as carbon sources
for survival. Acinetobacter sp. DL-28 has been shown to express the
novel enzyme, L-ribose isomerase (AcL-RbI), which catalyzes reversible
isomerization between L-ribose and L-ribulose. AcL-RbI showed the highest
activity to L-ribose, followed by D-lyxose with 47 % activity, and had no
significant amino acid sequence similarity to structure-known proteins,
except for weak homology with the D-lyxose isomerases from Escherichia
coli O157:H7 (18 %) and Bacillus subtilis strain (19 %). Thus,
AcL-RbI is expected to have the unique three-dimensional structure to
recognize L-ribose as its ideal substrate. The X-ray structures of AcL-RbI in
complexes with substrates were determined. AcL-RbI had a cupin-type beta-barrel structure, and the catalytic
site was found between two large beta-sheets with a bound metal ion. The
catalytic site structures clearly showed that AcL-RbI adopted a cis-enediol intermediate mechanism for the
isomerization reaction using two glutamate residues (Glu113 and
Glu204) as acid/base catalysts.
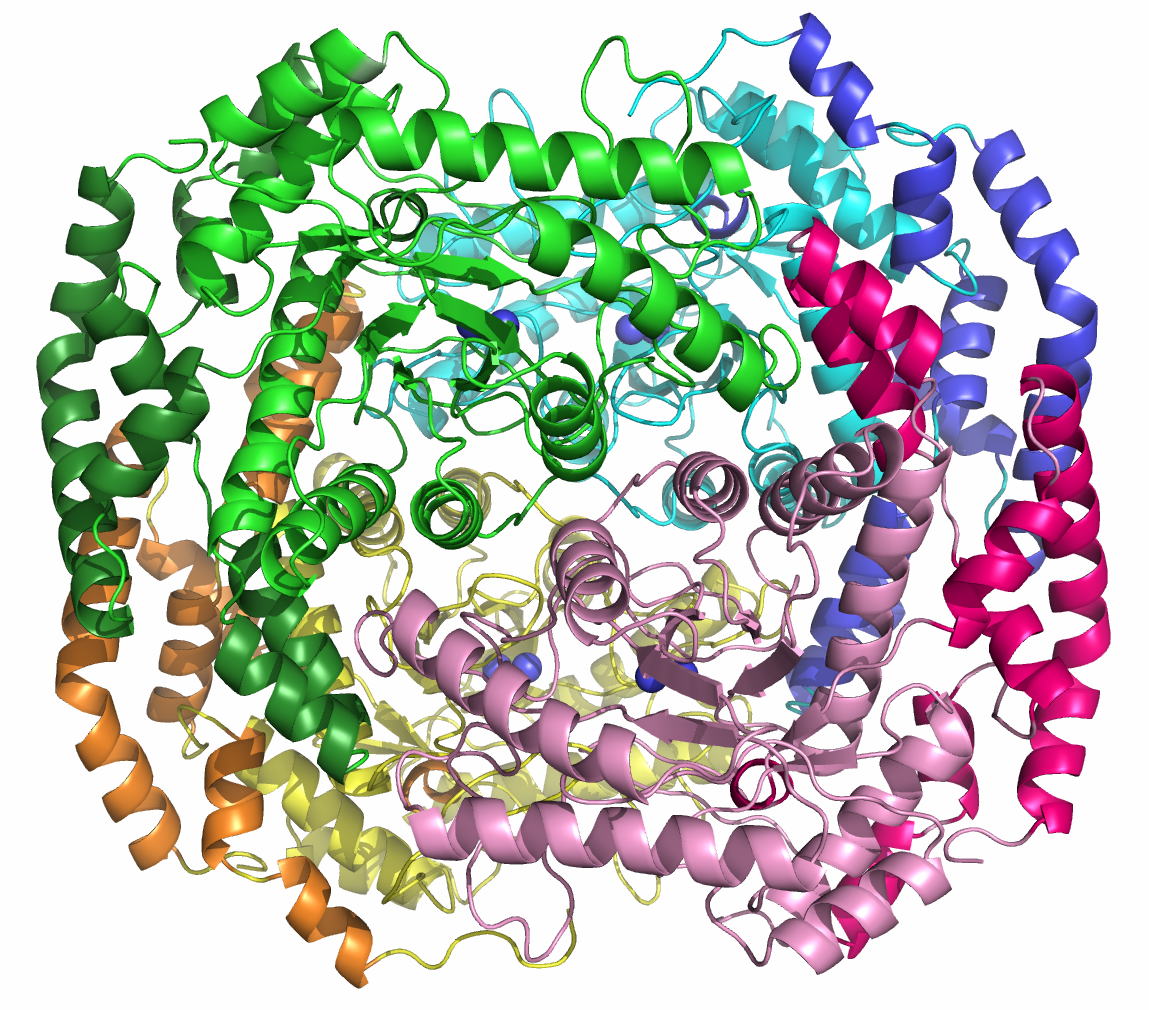
In its crystal form, AcL-RbI formed a unique homo-tetramer with many substrate
sub-binding sites, which likely facilitated capture of the substrate. FEBS J. (2014) 281, 3150-3164. |
|
|
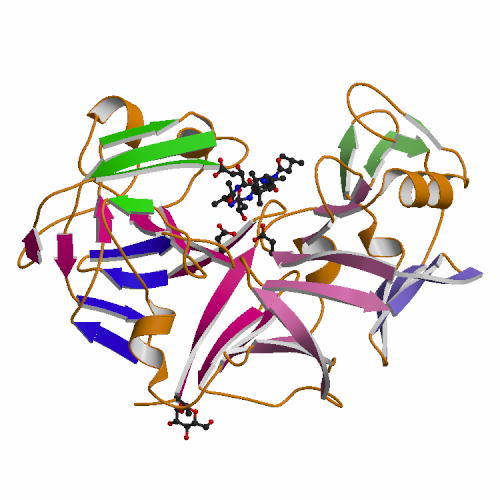
The
X-ray structures of Aspergillus oryzae
aspartic proteinase (AOAP) and its complex with inhibitor pepstatin
have been determined at 1.9 Å resolution. AOAP has a
crescent-shaped structure with two lobes (N-lobe and C-lobe) and the deep
active site cleft is constructed between them. At the center of the active
site cleft, two Asp residues (Asp33 and Asp214) form the active dyad with a hydrogen bonding solvent molecule between them. Pepstatin binds to the active site cleft via hydrogen
bonds and hydrophobic interactions with the enzyme. The structures of AOAP
and AOAP/pepstatin complex including interactions
between the enzyme and pepstatin are very similar
to those of other structure-solved aspartic proteinases and their complexes
with pepstatin. Generally, aspartic proteinases
cleave a peptide bond between hydrophobic amino acid residues, but AOAP can
also recognize the Lys/Arg residue as well as
hydrophobic amino acid residues, leading to the activation of trypsinogen and chymotrypsinogen.
The X-ray structure of AOAP/pepstatin complex and
preliminary modeling show two possible sites of recognition for the
positively charged groups of Lys/Arg residues
around the active site of AOAP. |
|
|
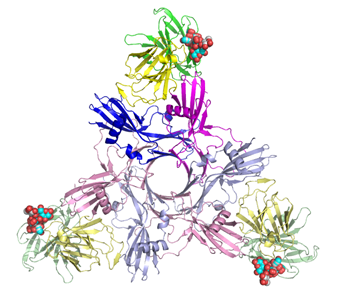
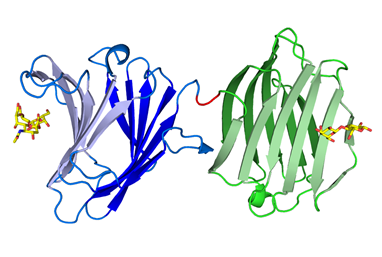
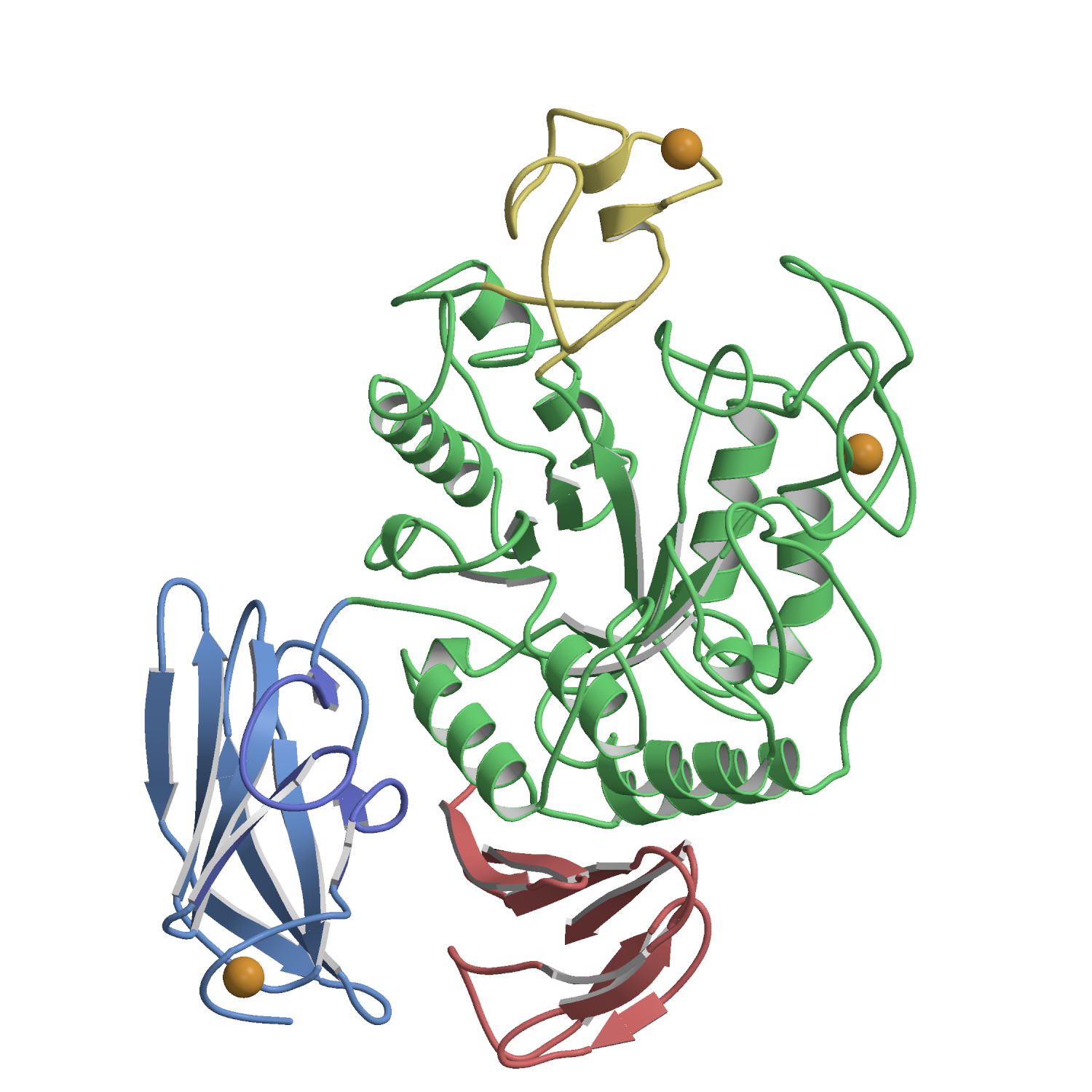
Clostridium
botulinum hemagglutinin (HA) subcomponent
in ) in complexes with sialylated oligosaccharides (4EN6, 4EN8) Clostridium
botulinum produces the botulinum neurotoxin, forming a
large complex as progenitor toxins in association with nontoxic nonhemagglutinin and/or several different hemagglutinin (HA) subcomponents, HA33, HA17 and HA70,
which bind to carbohydrate of glycoproteins from epithelial cells in the
infection process. To elucidate
the carbohydrate recognition mechanism of HA70, X-ray structures of HA70 from
type C toxin (HA70/C) in complexes with sialylated
oligosaccharides were determined, and a
binding assay by the glycoconjugate microarray
was performed. These results suggested that HA70/C can recognize both alpha-2-3- and alpha-2-6-sialylated
oligosaccharides, and that it has a higher affinity for alpha-2-3-sialylated
oligosaccharides. FEBS
Lett. (2012) 586, 2404-2410. |
|
|
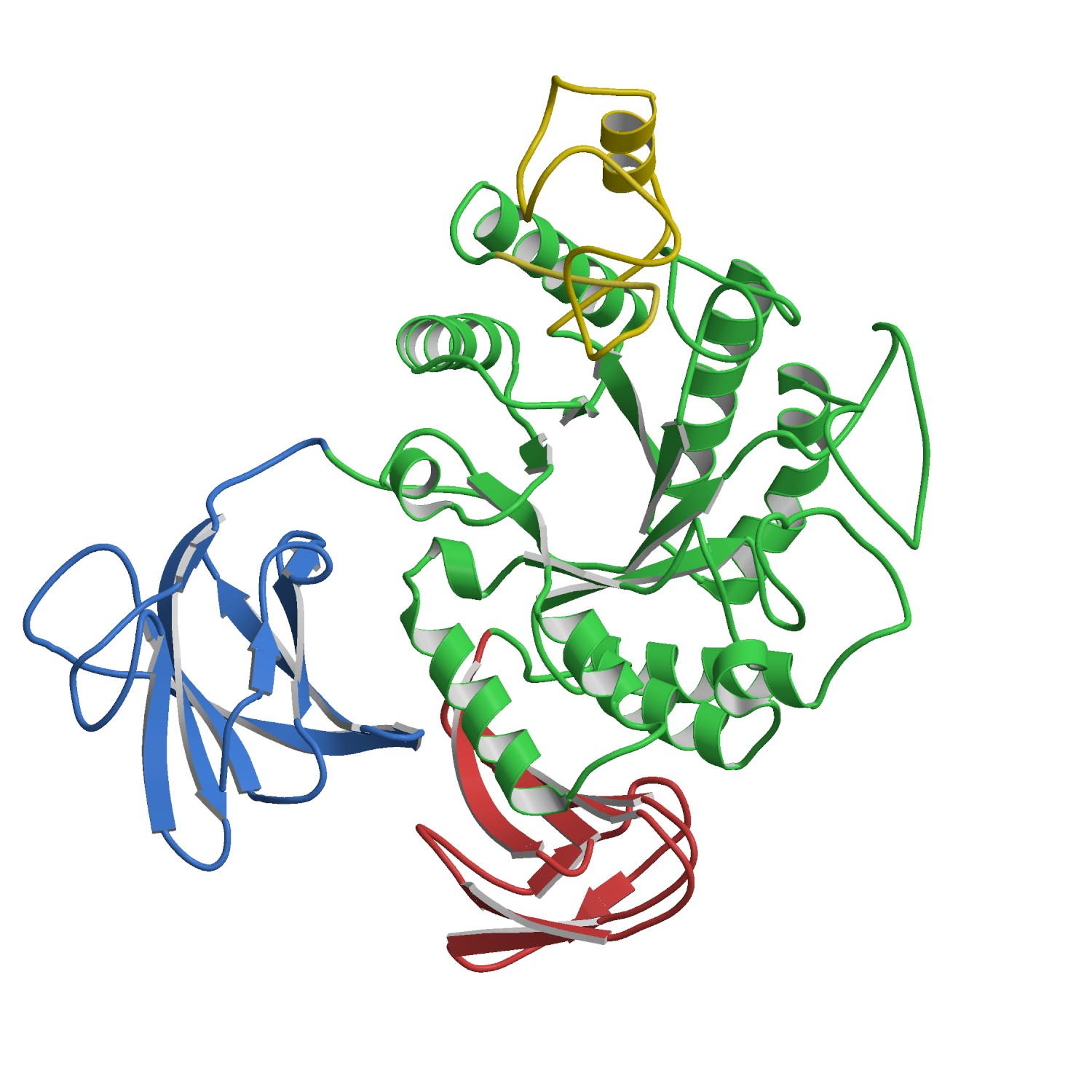
Endolysin (Psm) encoded by episomal phage phiSM101 of enterotoxigenic Clostridium perfringens
(4KRT) Gram-positive
bacteria possess a thick cell wall composed of a mesh polymer of
peptidoglycans, which provides physical protection. Endolysins encoded by
phages infecting bacteria can hydrolyze peptidoglycans in the bacterial cell wall, killing the host bacteria immediately.
The endolysin (Psm) encoded by episomal
phage phiSM101 of enterotoxigenic Clostridium perfringens
type A strain SM101 exhibits potent lytic activity towards most strains
of Clostridium perfringens.
Psm has an N-terminal catalytic domain highly
homologous to N-acetylmuramidases
belonging to the glycoside hydrolase 25 family, and C-terminal tandem
repeated bacterial Src homology 3 (SH3_3) domains
as the cell wall binding domain. The X-ray structure of full-length Psm and a catalytic domain of Psm
in complex with N-acetylglucosamine were determined to elucidate the catalytic reaction and cell wall recognition
mechanisms of Psm. The results
showed that Psm may have adopted a
neighboring-group mechanism for the catalytic hydrolyzing reaction in which
the N-acetyl carbonyl group of the
substrate was involved in the formation of an oxazolinium
ion intermediate. Based on structural comparisons with other endolysins and a modeling study, we proposed that tandem
repeated SH3_3 domains of Psm recognized the
peptide side chains of peptidoglycans to assist the catalytic domain
hydrolyzing the glycan backbone. Molecular
Microbiology (2014) 92, 326–337. |
|
|
Human
galectin-8 in a protease-resistant mutant form (3VKM) Galectin-8 is a
tandem-repeat-type beta-galactoside-specific animal
lectin possessing N-terminal and C-terminal carbohydrate recognition domains
(N-CRD and C-CRD, respectively), with a difference in carbohydrate binding
specificity, involved in cell–matrix
interaction, malignant transformation, and cell adhesion. N-CRD shows strong
affinity for alpha-2–3-sialylated
oligosaccharides, a feature unique to galectin-8. C-CRD usually shows lower
affinity for oligosaccharides but higher affinity for N-glycan-type branched
oligosaccharides than does N-CRD. There have been many structural studies on galectins with a single carbohydrate recognition domain
(CRD), but no X-ray structure of a galectin
containing both CRDs has been reported. Here, the X-ray structure of a
protease-resistant mutant form of human galectin-8 possessing both CRDs and
the novel pseudodimer structure of galectin-8 N-CRD
in complexes with alpha-2–3-sialylated
oligosaccharide ligands were determined. The results revealed a difference in
specificity between N-CRD and C-CRD, and provided new insights into the
association of CRDs and/or molecules of galectin-8. FEBS J. (2012) 279, 3937-3951. |
|
|
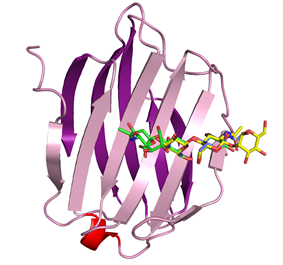
Human
galectin-9 C-terminal domain (3NV3) The galectins are a family of beta-galactoside-specific
animal lectins which contain conserved elements for carbohydrate recognition,
and have attracted much attention as novel regulators of physiological
systems. Currently, there are 14 members of the mammalian galectin
family, classified into three subtypes on the basis of structure; the
prototype, the chimera-type, and the tandem-repeat-type galectins.
Human galectin-9, having high affinity for N-glycan-type oligosaccharides with branches and sialylated oligosaccharides, is involved in eosinophil chemoattraction and apoptosis of T helper type 1 cells, in the immune sysytem. To
elucidate this unique feature, X-ray structures of human galectin-9
C-terminal domain in complexes with the bianntenary
pyridylaminated oligosaccharide and alpha-2-3 sialyllactose were determined. J. Biol. Chem. (2010) 285, 36969-36976. |
|
|
Iba1
(ionized calcium-binding adaptor molecule 1) with 147 amino acid residues has
been identified as a calcium (Ca2+)-binding protein, expressed
specifically in microglia/macrophages, and is expected to be a key factor in
membrane ruffling which is a typical feature of activated microglia. We have
determined the crystal structure of human Iba1 in Ca2+-free form and
mouse Iba1 in Ca2+-bound form, to a resolution of 1.9 Å and 2.1 Å,
respectively. X-ray structures of Iba1 revealed a compact, single-domain
protein with two EF-hand motifs, showing similarity in overall topology to
partial structures of the classical EF-hand proteins troponin C and calmodulin. In mouse Iba1, the second EF-hand contains a
bound Ca2+, but the first EF-hand does not, which is often the
case in S100 proteins, suggesting that Iba1 has S100 protein-like EF-hands.
The molecular conformational change induced by Ca2+-binding of
Iba1 is different from that found in the classical EF-hand proteins and/or
S100 proteins, leading to the formation of a dimer in crystals, which
demonstrates that Iba1 has a novel molecular switching mechanism dependent on
Ca2+-binding, to interact with target molecules. J. Mol. Biol. (2006) 364, 449-457. |
|
|
J.
Mol. Biol. (2007) 374, 443-453. |
|
|
J. Mol. Biol. (2007) 365, 1505-1516. |
|
|
The
X-ray structures of red yeast Sporobolomyces salmonicolor carbonyl reductase (SSCR) and its complex
with a coenzyme, NADPH, have been determined at a resolution of 1.8 Å and 1.6
Å, respectively. SSCR has two domains, an NADPH-binding domain and a
substrate-binding domain, and belongs to the short-chain
dehydrogenases/reductases family. The structure of the NADPH-binding domain
and the interaction between the enzyme and NADPH are very similar to those
found in other structure-solved enzymes belonging to the short-chain
dehydrogenases/reductases family, while the structure of the
substrate-binding domain is unique. SSCR has stereoselectivity
in its catalytic reaction, giving rise to excessive production of
(S)-alcohols from ethyl 4-chloro-3-oxobutanoate. The X-ray structure of the
SSCR/NADPH complex and preliminary modeling show that the formation of the
hydrophobic channel induced by the binding of NADPH is closely related to the
stereoselective reduction by SSCR. J. Mol. Biol. (2005) 352, 551-558. |
|
|
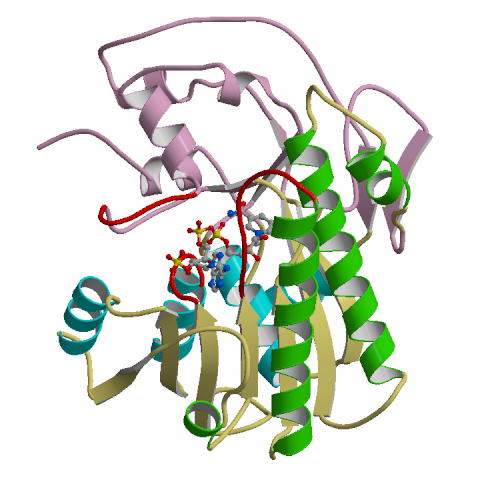
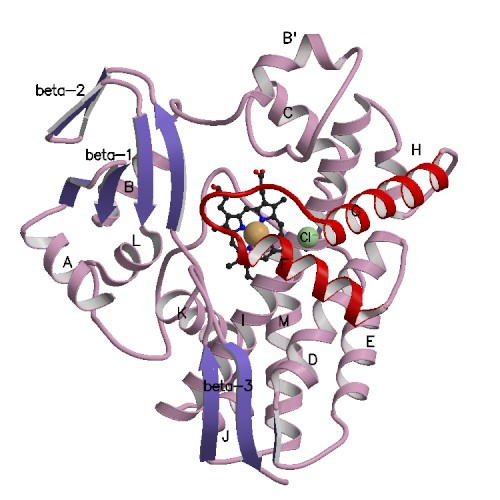
A
cytochrome P450 from acidothermophilic archaebacterium Sulfolobus tokodaii strain7. S. tokodaii
strain7 (P450st) carrying histidine6-tag has been expressed in E. coli and
purified with high yield and homogeneity. The X-ray structure of P450st was
determined at 3.0 Å resolution. Structural
comparison with cytochrome P450 from Sulfolobus solfataricus (CYP119)
suggests that the region from the F to G helices and the binding Cl- is
possibly responsible to the affinity of a ligand coordinating to the heme iron. The direct electrochemistry of P450st in a
DDAB film on the PFC electrode has been also demonstrated. The
quasi-reversible redox response has been observed even at 80 °C. J. Inorg. Biochem. (2004) 98, 1194-1199. |
|
|
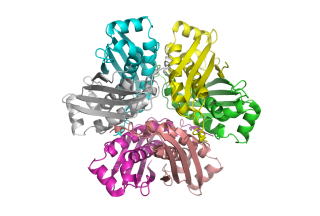
The crystal
structure of a putative molybdenum-cofactor (Moco)
biosynthesis protein C (MoaC) from Sulfolobus tokodaii (ST0472)
was determined at 2.2 A resolution. The crystal belongs to the monoclinic
space group C2, with unit-cell parameters a = 123.31, b = 78.58, c = 112.67
A, beta = 118.1 degrees . The structure was solved
by molecular replacement using the structure of Escherichia coli MoaC as the probe model. The asymmetric unit is composed
of a hexamer arranged as a trimer of dimers with noncrystallographic 32 symmetry. The structure of
ST0472 is very similar to that of E. coli MoaC;
however, in the ST0472 protein an additional loop formed by the insertion of
seven residues participates in intermonomer
interactions and the new structure also reveals the formation of an interdimer beta-sheet. These features may contribute to
the stability of the oligomeric state. Acta Crystallogr. Sect F. (2008) 64, 589-592. |
|
|
A
56-kDa selenium-binding protein (SBP56) was originally discovered as a cytosolic
protein with the selenium-binding activity (Bansal et al., 1989). SBP56 is a
highly conserved protein found widely in microorganisms,
plants and animals. SBP56 was reported to be
involved in the transport of selenium compounds and intra-Golgi transport,
however, its function in microorganisms has been still unclear. To
obtain new insights into structure-function relationship of SBP56, we have
determined the X-ray structure of Sulfolobus tokodaii hypothetical SBP56
(ST0059), which has 39 % identity in amino acid sequence with human SBP56. |
|
|
|
|
|
|
|